
U. Faysal Ilhan
1305LGRT
ilhan@chem.umass.edu
(413) 545-4865
Education:
Ph.D. University of Massachusetts, MA; 1996-
B.S Bogazici University -Istanbul 1996 (Honor)
Research Interests:
Polymer chemistry, organic synthesis, materials.
Research:
1) Recognition-Mediated Unfolding of a Self-Assembled Polymeric Globule
I am currently working on the creation of "plug and play" polymers that use non-covalent interactions to modify polymer properties post-synthetically. In preliminary research, we have synthesized a soluble, diaminotriazine-functionalized polymer 1. This polymer adopts a micelle-like structure in chloroform solution, driven by the formation of intramolecular hydrogen bonds between the triazine moieties. This polymer can be unfolded via non-specific competition from solvents, as well as through specific recognition using a complementary receptor.
Preliminary prediction of the structure of polymer 1 was obtained through molecular dynamics calculations on a model polymer, poly(styrene--p-(methyldiaminotriazine) styrene (Figure 1). In vacuo calculations predicted a highly compact structure containing multiple intramolecular hydrogen bonds between triazine sidechains. This compact structure was quite robust, maintaining integrity at elevated temperatures. Direct confirmation of the compact micellar folding behavior of polymer 1 in non-competitive solvents was obtained through gel permeation chromatography (GPC) experiments in CHCl3.
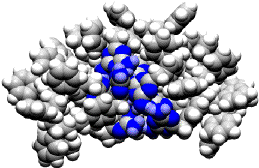
Figure 1. Micelle-like structure predicted (Amber forcefield) for atactic polystyrene (40 monomer units total) with methylenediaminotriazine substitution at every fourth carbon.
To quantify the thermodynamics of folding and recognition-mediated unfolding, we performed variable temperature 1H NMR titrations between flavin 3 and polymer 1 as well as monomer 2. To get a further insight into the entropic balance achieved by this system, we used thermodynamic values obtained from NMR studies providing a cycle for the recognition-controlled micellar folding/unfolding process (Figure 2). From the thermodynamic cycle, we see that there is an almost perfect balance of entropics for the recognition and unfolding processes, resulting in the very weak temperature dependence for the polymer 1-flavin 3 complex. The large increase in entropy associated with the unfolding process indicates the complex is quite rigidly self-assembled, consistent with the GPC results in CHCl3. This hypothesis is supported by the relatively large enthalpic cost of unfolding: the enthalpy of the unfolding process (1uÆ1f) is roughly two-thirds that of the recognition process (1u+3Æ1u.3), where three hydrogen bonds are formed. This indicates that there are approximately two dynamic hydrogen bonds per triazine in the folded state.
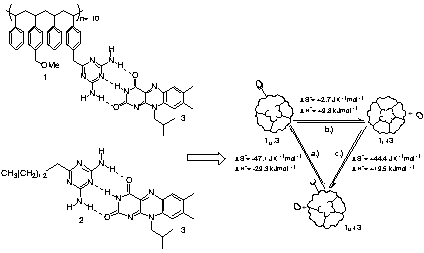
Figure 2. Polymer 1-flavin 3 and monomer 2-flavin 3 complexes and thermodynamic cycle for polymer 1-flavin 3 complexation.
Applications of diaminotriazine functionalized polystyrene
a) Recognition and encapsulation of an electroactive guest:
The engineered arrangement of converging functionality within polymer 1, provides a potential means for encapsulation of guests through specific non-covalent interactions. We applied this strategy to the structural and functional site isolation of electroactive guest 6-ferrocenyluracil 2 within the core of globular polymer 1 (Figure 3). To show the site-isolation of electroactive target 2, we have performed NMR and electrochemical studies.
We have obtained an association constant of 487±52 M-1 for polymer 1/guest 2 complexation through NMR titration studies. This figure corresponds to an over 13-fold enhancement in binding relative to that obtained for flavin 4 / polymer 1 system (36 M-1). Such a dramatic change requires 4 point H-bonding on guest 2, compared to 3 point H-bonding in flavin 4 case. Polymer needs to wrap around the guest 2 to get the mentioned type of binding.
Functional demonstration of site isolation in the guest 2-polymer 1 complex was obtained by using cyclic voltammetry. The voltammogram of ferrocene derivative 2 displays a sharp reduction peak arising from precipitation of the charged, oxidized ferrocenium species. Addition of one equivalent of monomer 3 to the solution of 2 had minimal affect on the electrochemical behavior, with only a slight decrease in the peak current of the reduction couple observed. In contrast, when an equimolar quantity of polymer 1 was added to a solution of 2, the voltammogram of 2 became almost completely reversible. This demonstrates that encapsulation of guest 2 within polymer 1 effectively prevents aggregation the oxidized species.
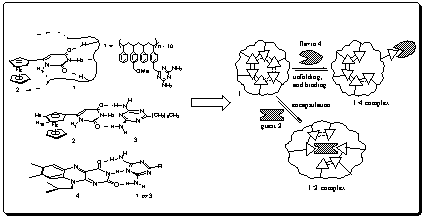
Figure 3. Illustration showing difference in macroscopic conformational reorganization of polymer 1 during encapsulation and binding processes for the 2.1 and 4.1 systems, respectively.
b) Self assembly of nanoparticles:
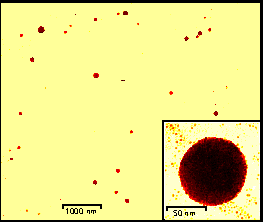
Figure 4. TEM micrograph of gold colloid-polymer 1 aggregates; inset: representative self assembled microparticle.
We have utilized diaminotriazine functionalized polymer 1, to provide a general means for the controlled self-assembly of nanoparticles. In this strategy, colloidal gold particles functionalized with recognition elements serve as the "bricks", while polymers bearing complementary functionality serve as "mortar", holding together the colloidal particles. Using this strategy, the conformational flexibility of the polymer compensates for irregularities in the size and shape of the aggregate structure, allowing the creation of spherical arrays. According to our SAXS and TEM investigations, monolayer protected 2 nm gold particles self-assemble into discrete 97±17 nm spherical microparticles, each comprised of between 3000 and 7000 individual subunits (Figure 4).
2) Control of Polymer Solution Structure via Intra- and Intermolecular Aromatic Stacking
In another research, we have synthesized anthracene-functionalized polymer 1 which folds into a compact globular structure in nonpolar media. This folding arises from intramolecular aromatic-aromatic interactions of the anthracene sidechains (Figure 5). Also we have performed variable temperature GPC and fluorescence studies between polymer 1 and an external guest picric acid 2. Polymer 1 binds strongly to picric acid 2, through donor-acceptor electrostatic interactions between the electron-rich anthracene sidechains and the electron-deficient picric acid guests. We have shown that this strong binding stabilizes the thermally-labile folded conformation of polymer 1, dramatically altering the temperature dependence of polymer unfolding.
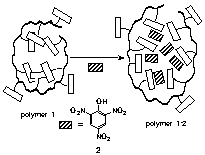
Figure 5. Schematic diagram of polymer 1-picric acid 2 host-guest complexation.
3) Kinetic Trapping of Host-Guest Complexes in a Polymeric Matrix
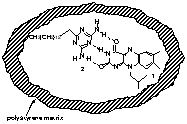
Figure 6. Schematic illustration of isolated flavin 1-triazine 2 host-guest complexes in polystyrene matrix.
Polymer matrix isolation is an effective technique for the isolation and immobilization of molecules. Application of this methodology to the isolation of host-guest assemblies is hampered by issues of competition and aggregation. Matrix formation from polar polymers creates a competitive environment, disrupting the desired interactions such as hydrogen bonding. Conversely, creation of matrices using non-polar polymers can cause aggregation and concomitant phase separation of polar host-guest complexes. To overcome the issue of phase separation in non-polar polar matrices, we have explored methods of trapping host-guest complexes. We kinetically isolated both individual host flavin 1 and host flavin 1 &endash; guest triazine 2 complex in a highly non-polar polystyrene matrix through spin-casting of polymer solutions. Self-aggregation of polar flavin 1 in these non-polar polymer films was prevented by adjusting the ratio between polystyrene and flavin 1 in solution prior to spin casting. We performed fluorescence studies on these spin-cast films to quantify the host-guest complexation in the polystyrene matrix.
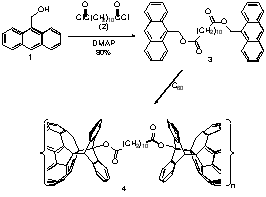
4) Thermoreversible Polymerization. Formation of Fullerene-Diene Oligomers and Copolymers
I am also using Diels-Alder methodology to synthesize fullerene-based copolymers. Diels-Alder reaction of C60 with bis-anthracene 3 provides linear copolymer 4. This material is stable at room temperature, but undergoes reversion to the monomeric species upon heating. This thermal depolymerization process was observed at temperatures above 60°C, with an activation energy of 17.1 kcal/mol. This process can be cycled multiply without degradation of the reactive functionalities. In addition to its thermal properties, this polymer retains the rich redox chemistry of the fulleroids. Electrochemical and EPR studies demonstrate copolymer 4 undergoes multiple reductions without decomposition to monomeric species.
Publications:
6) "Kinetic Trapping of Host-Guest Complexes in a Polymeric Matrix" F. Ilhan, L. Diamondis, L. Gautreau, V. Rotello, J. Chem. Soc., Chem. Comm., in press.
5) "Self-Assembly of Nanoparticles into Giant Spherical Arrays" A. Boal, F. Ilhan, J. DeRouchey, T. Thurn-Albrecht, T. Russell, V. Rotello, Nature, in press.
4) "Encapsulation of an Electroactive Guest in a Dynamically Self-Assembled Polymer" T. Galow, F. Ilhan, G. Cooke. V. Rotello, J. Am. Chem. Soc., in press.
3) "Control of Polymer Solution Structure via Intra- and Intermolecular Aromatic Stacking" F. Ilhan, M. Gray, K. Blanchette, V. Rotello, Macromolecules, 1999, 32, 6159-6162.
2) "Recognition-Mediated Unfolding of a Polymeric Globule" R. Deans, F. Ilhan, V. Rotello, Macromolecules. 1999, 32, 4956-4960.
1) "Thermoreversible Polymerization. Formation of Fullerene-Diene Oligomers and Copolymers" F. Ilhan, V. Rotello, J. Org. Chem., 1999, 64, 1455-1458.
Presentations:
3) Control of polymer structure and function through intramolecular self-assembly. Rotello V, Ilhan F ABSTR PAP AM CHEM S 218: U39-U40 Part 2 AUG 22 1999.
2) Thermoreversible materials based on fullerene Diels-Alder methodology. Rotello V, Ilhan F. ABSTR PAP AM CHEM S 218: U155-U156 Part 2 AUG 22 1999.
1) Plug and play polymers. Diversity through supramolecular processes. Ilhan UF, Rotello VM ABSTR PAP AM CHEM S 216: U393-U393 Part 2 AUG 23 1998.
Return to group members page
Return to group homepage