Research Overview - Craig T. Martin
Transcription lies at the heart of cellular regulation, and understanding the complex machinery involved in the synthesis of RNA is critical to understanding that regulation. We now have crystal structures for eukaryotic, prokaryotic, and phage RNA polymerases, but many questions remain regarding the mechanisms of the individual phases of transcription. Indeed, regulation occurs not only at the initial selection of the promoter, but also at the initial synthesis step, during the initial movement away from the promoter, during elongation, and at sequence-specific termination. The structure-function relationships underlying each of these processes are key to understanding transcription.
T7 RNA polymerase is the simplest and best-understood of the RNA polymerases. In a sense, it is the core of an RNA polymerase. As such, it represents an ideal model system in which to study transcription.
Fluorescence probes of melting. In order to follow the progress of the melted bubble during the various stages of transcription, we have used the site-specific incorporation of fluorescent nucleotide analogs into the DNA. In the summary below, dark circles represent fluorophores stacked within a duplex, while lighter circles indicate the increase in fluorescence associated with melting of the DNA bubble.

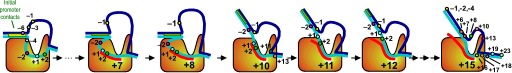
The initially melted bubble is about 8 bases in size [xx]. As transcription initiates, the upstream edge of the bubble remains fixed as the downstream edge expands [xx]. Just past the synthesis of an 8 base RNA, the upstream edge of the bubble collapses, ultimately returning the bubble to the constant 8 base size characteristic of an elongation complex [33], clear of the promoter.
A large structural change in the protein. The structures above reveal a very large structural rearrangement of about 1/3 of the protein. Based on topological and biochemical considerations, we have developed a movie to illustrate how we think this transition occurs.
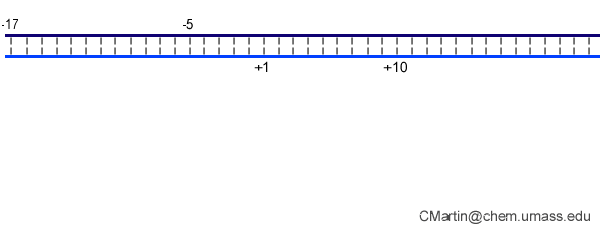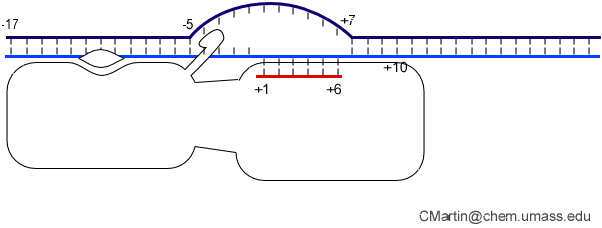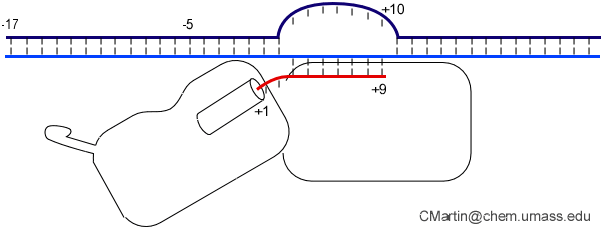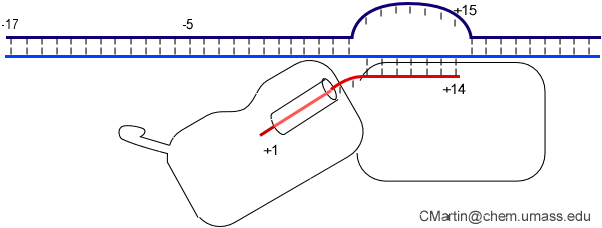
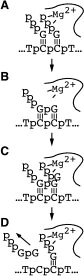
Biochemical studies complement the fluorescence studies, revealing precisely when promoter contacts are lost and when the 5' end of the RNA is displaced from the template. The cartoon above shows details of a functional model we have proposed (see also our animation combining the structural and biochemical models). In particular, we argue that binding of the promoter DNA drives the Val loop into the DNA, initiating bubble formation.
the structural movie shows that the promoter-protein contacts can be retained (and therefore the bubble can remain open) through translocation to about position +8, consistent with our biochemical studies. At that point, however, the specificity loop is pulled from the promoter-binding platform, releasing the DNA. This in turn, removes the Val loop from the upstream edge of the bubble, allowing collapse. Collapse then helps to drive initial displacement of the 5' end of the RNA, leading it into the just-formed RNA exit channel. Threading of the RNA into the channel signals the formation of a stable elongation complex.
The following describes recent advances from our laboratory.
View animated cartoon in:
QuickTime
|
Flash
|
GIF format
Abortive cycling and promoter clearance. Our very recent work has turned towards understanding the RNA polymerase as it transcribes away from the promoter. Fluorescent base analogs have proven extremely valuable in this analysis and we have extended our earlier characterization of the initial promoter complex to provide an accurate mapping of the downstream edge of the bubble [unpublished]. Additionally, we have shown that in the synthesis of a trinucleotide transcript, the downstream edge of the bubble does not move the active site approaches very close to the downstream edge of the melted region, a result
Available crystal structures are consistent with earlier footprinting studies in that the RNA polymerase can transcribe at least 3-6 bases without releasing the upstream duplex promoter contacts. Modeling from the crystal structure, however, predicted that during this early phase, the heteroduplex can be no longer than 3 base pairs. Using fluorescent base analogs, we have mapped the collapse of the initial bubble and the extent of heteroduplex formation [37]. The results show very clearly that the initial heteroduplex grows to a maximal length of about 10 base pairs. On translocation from 8 base pairs to 9 or 10, the initial melted bubble collapses, indicating the loss of promoter contacts, and on translocation from position +10 to +11, the initially synthesized RNA first begins to peel away from the heteroduplex. These results have important implications on the mechanism driving the structural changes occurring at this unique stage in transcription.
Probe development. A necessary component of the above analyses was the advent of fluorescent base analogs beyond the initial analog of adenine, 2aminopurine. In the above work, we have used two new analogs: 6MI, an analog of G (collaborative with M. Hawkins, NCI) and pyrroloC, an analog of C (in cooperation with J. Randolph and H. Mackey, Glen Research Corp.), all of which show duplex-enhanced fluorescence quenching). This has extended the sequence contexts which can be probed (a T analog, furanoT, will be characterized in the near future). These new probes have excitation maxima more distant from protein absorbances, reducing greatly complications of background fluorescence. We are currently collaborating with J. B. A. Ross (Mt. Sinai) to use lifetime measurements as a means of characterizing translocational heterogeneity in various stalled complexes, to clarify further the picture above. Finally, we have demonstrated the use of mismatch probes to clearly distinguish DNA:DNA from RNA:DNA duplexes [37]. These new tools and approaches will likely be of utility in a wide variety of protein-nucleic acid systems.
Site-specific de novo initiation. Template strand DNA directs the synthesis of RNA and is clearly essential for polymerase function, however, we have demonstrated that start site selection is not strongly dependent on the precise nature of the coupling between the non-transcribed and the transcribed regions of the template strand [29]. This has led to more recent studies (with W. T. McAllister) which strongly favor a model in which the linkage to the upstream domain serves merely as a tether to increase the local concentration of an appropriate initiation sequence near the active site [35], despite the appearance in the crystal structure of what appears to be active guidance by the linking DNA.
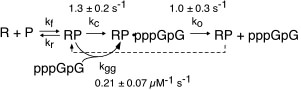
 Kinetic studies of initiation. As described above, our early work used steady state kinetics to assess functional structural elements of promoter binding. We subsequently turned our attention to pre-steady state kinetic analyses of initial dinucleotide synthesis in order to elucidate the mechanism of initiation [32]. Fitting kinetic data to integrated rate equations for specific, complex mechanisms, we have demonstrated that dinucleotide re-binding (product inhibition in this initial assay) must be included in any such mechanism and that substrate activation plays an important role in mediating product release. These results present an essential foundation to mechanistic studies of translocation through the initial abortive region, leading to promoter clearance.
Kinetic studies of initiation. As described above, our early work used steady state kinetics to assess functional structural elements of promoter binding. We subsequently turned our attention to pre-steady state kinetic analyses of initial dinucleotide synthesis in order to elucidate the mechanism of initiation [32]. Fitting kinetic data to integrated rate equations for specific, complex mechanisms, we have demonstrated that dinucleotide re-binding (product inhibition in this initial assay) must be included in any such mechanism and that substrate activation plays an important role in mediating product release. These results present an essential foundation to mechanistic studies of translocation through the initial abortive region, leading to promoter clearance.
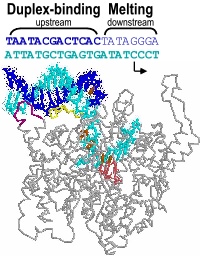 Promoter melting. Two unique roles of an RNA polymerase are promoter melting and the de novo (without a primer) initiation of RNA synthesis. With respect to the former question, we have used fluorescent base analogs to characterize the initially melted bubble, mapping the melted region (the second domain described above) and demonstrating that promoter melting is extremely rapid, suggesting that melting occurs coincident with initial promoter binding [28]. The latter result was expanded in thermodynamic measures of the binding of full length and truncated promoter constructs [30]. The observation that truncated DNA constructs representing the duplex binding domain (only) bind to the protein more strongly than full length DNA led us to a model in which the unfavorable process of melting is obligatorily coupled to binding. Our gel-based bending assays [31] support this notion and have suggested a structural mechanism in which binding-induced bending of the DNA leads to melting, a model which we are currently testing by site-directed mutagenesis.
Promoter melting. Two unique roles of an RNA polymerase are promoter melting and the de novo (without a primer) initiation of RNA synthesis. With respect to the former question, we have used fluorescent base analogs to characterize the initially melted bubble, mapping the melted region (the second domain described above) and demonstrating that promoter melting is extremely rapid, suggesting that melting occurs coincident with initial promoter binding [28]. The latter result was expanded in thermodynamic measures of the binding of full length and truncated promoter constructs [30]. The observation that truncated DNA constructs representing the duplex binding domain (only) bind to the protein more strongly than full length DNA led us to a model in which the unfavorable process of melting is obligatorily coupled to binding. Our gel-based bending assays [31] support this notion and have suggested a structural mechanism in which binding-induced bending of the DNA leads to melting, a model which we are currently testing by site-directed mutagenesis.
 Promoter recognition and binding. Our early research (1988-1996) employed functional group substitutions in DNA to characterize promoter contacts and interactions in the T7 RNA polymerase model system [21, 22, 25, 27]. The results are consistent with a two-domain description of the promoter and predicted energetically critical major groove contacts in the central part of the proposed upstream duplex binding region of the promoter, with less important minor groove contacts in the far upstream part of the bound duplex. Our results further showed that within the melted domain encompassing the start site for transcription, the nontemplate strand provides few energetically important contacts [23]. These results are fully consistent with the subsequent (1999) crystal structure of the promoter bound complex (and with a large body of full base pair substitution measurements), and provide energetic information not available from the structure alone.
Promoter recognition and binding. Our early research (1988-1996) employed functional group substitutions in DNA to characterize promoter contacts and interactions in the T7 RNA polymerase model system [21, 22, 25, 27]. The results are consistent with a two-domain description of the promoter and predicted energetically critical major groove contacts in the central part of the proposed upstream duplex binding region of the promoter, with less important minor groove contacts in the far upstream part of the bound duplex. Our results further showed that within the melted domain encompassing the start site for transcription, the nontemplate strand provides few energetically important contacts [23]. These results are fully consistent with the subsequent (1999) crystal structure of the promoter bound complex (and with a large body of full base pair substitution measurements), and provide energetic information not available from the structure alone.
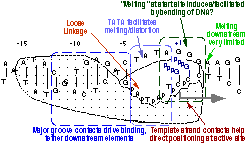 We have confirmed a central "core" within the promoter which is responsible for tight binding to the RNA polymerase. Energy from this interaction appears to be used to drive melting of the DNA near the start site. Other recent results have yielded unexpected (and therefore very intersting!) results, providing information on how the polymerase directs the initiating bases of the DNA template strand into the protein active site. Click here (or on the picture above) for more detailed information.
We have confirmed a central "core" within the promoter which is responsible for tight binding to the RNA polymerase. Energy from this interaction appears to be used to drive melting of the DNA near the start site. Other recent results have yielded unexpected (and therefore very intersting!) results, providing information on how the polymerase directs the initiating bases of the DNA template strand into the protein active site. Click here (or on the picture above) for more detailed information.